Quirónsalud








Blog del Dr. Daniel Martín Fernández-Mayoralas. Neurología. Complejo Hospitalario Ruber Juan Bravo y Olympia Centro Médico Pozuelo
- 20255mar
SYNGAP1: entendiendo un Gen Clave en el Neurodesarrollo
Hoy quiero hablarles sobre un gen fascinante y, a la vez, crucial para el desarrollo neurológico de nuestros niños: el gen SYNGAP1. Aunque no sea un nombre que escuchemos a menudo, su papel es fundamental en la forma en que el cerebro se conecta y funciona.

¿Qué es el gen SYNGAP1?
Imaginen que el cerebro es una ciudad muy grande, con miles de calles y avenidas donde la información viaja constantemente. El gen SYNGAP1 es como un gerente de tráfico esencial en esas intersecciones, asegurando que la información fluya de manera eficiente. Este gen produce una proteína llamada SynGAP1, que se encuentra en las sinapsis, que son las conexiones entre las neuronas. Esta proteína ayuda a regular la fuerza de estas conexiones, lo que es vital para el aprendizaje, la memoria y el desarrollo cognitivo.
¿Qué ocurre cuando SYNGAP1 no funciona correctamente?
Cuando hay una mutación o un error en el gen SYNGAP1, la proteína que produce no puede realizar su trabajo correctamente. Esto puede llevar a una serie de desafíos en el desarrollo neurológico, conocidos como el trastorno relacionado con SYNGAP1. Los síntomas pueden variar de persona a persona, pero algunos de los más comunes incluyen:
- Retraso en el desarrollo: los niños pueden tardar más en alcanzar hitos importantes como gatear, caminar o hablar.
- Discapacidad intelectual: dificultades en el aprendizaje y la comprensión.
- Epilepsia: crisis epilépticas, a menudo de diferentes tipos, que pueden ser difíciles de controlar.
- Trastorno del espectro autista (TEA): dificultades en la interacción social y la comunicación, así como comportamientos repetitivos.
- Problemas de comportamiento: como hiperactividad, impulsividad o irritabilidad.
- Trastornos del sueño: dificultad para conciliar el sueño o mantenerlo.
- Ataxia: problemas de coordinación y equilibrio.
¿Qué hemos aprendido recientemente sobre SYNGAP1?
Un estudio reciente analizó datos de muchos pacientes con el trastorno relacionado con SYNGAP1, recopilados a través de un registro digital. Este estudio confirmó hallazgos previos sobre la importancia del gen y añadió nueva información valiosa:
- Epilepsia: el estudio encontró que la epilepsia es muy común en personas con el trastorno relacionado con SYNGAP1, y a menudo comienza después de que se identifica un retraso en el desarrollo. Algunos pacientes experimentan tipos específicos de crisis epilépticas, como las ausencias atípicas o las crisis atónicas.
- Problemas de comportamiento: se observó que los problemas de comportamiento son más comunes en aquellos que también tienen problemas de sueño y ansiedad, lo que subraya la importancia de abordar estos problemas de manera integral.
- Variantes genéticas: se identificaron algunas diferencias en los síntomas dependiendo de la ubicación específica de la mutación en el gen SYNGAP1. Por ejemplo, las personas con mutaciones en ciertas partes del gen pueden tener más probabilidades de desarrollar la capacidad de hablar en frases.
- Tratamientos: el estudio también proporcionó información sobre los medicamentos que se utilizan con mayor frecuencia para tratar la epilepsia, los problemas de sueño y los problemas de comportamiento en personas con el trastorno relacionado con SYNGAP1. Un diagnóstico temprano puede llevar a intervenciones más tempranas, como terapia del habla, terapia ocupacional y manejo de las convulsiones. Además, el apoyo emocional y la conexión con otras familias que están pasando por lo mismo pueden ser de gran ayuda.
El futuro de la investigación en SYNGAP1
La investigación sobre SYNGAP1 está avanzando rápidamente. Los científicos están trabajando para comprender mejor cómo este gen afecta el cerebro y cómo podemos desarrollar terapias más efectivas.
En resumen
El gen SYNGAP1 es una pieza clave en el rompecabezas del desarrollo neurológico. Aunque el trastorno relacionado con SYNGAP1 puede presentar desafíos significativos, la investigación continua y una mejor comprensión del gen nos dan esperanza para el futuro. Espero que este post haya sido útil para entender mejor el gen SYNGAP1. ¡Gracias por leer!
BIBLIOGRAFÍA: Wiltrout K, Brimble E, Poduri A. Comprehensive phenotypes of patients with SYNGAP1-related disorder reveals high rates of epilepsy and autism. Epilepsia. 2024;00:1–11. https://doi.org/10.1111/epi.17913
Gen SYNGAP1 - neurodesarrollo - retraso en el desarrollo - discapacidad intelectual - epilepsia - trastorno del espectro autista - TEA - hiperactividad - impulsividad - irritabilidad - trastornos del sueño - ataxia - neuropediatra - Quirónsalud - Hospital Universitario Ruber Juan Bravo - Hospital Universitario Quirónsalud Madrid - Dr. Daniel Martín Fernández-Mayoralas2 comentarios - 202423sep
Síndrome X frágil

¿Qué es el síndrome X frágil?
El síndrome de X frágil (FXS) es un trastorno genético neurológico, ligado al cromosoma X, que afecta a la capacidad intelectual y el comportamiento. Se trata de la causa hereditaria más común de discapacidad intelectual y afecta a varones y mujeres, pero de forma diferente.
¿A qué edades se suele diagnosticar?
En los varones suele presentarse como un retraso psicomotor desde las primeras edades, asociada a una hipotonía generalizada de leve a moderada. El fenotipo, esto es, el aspecto físico, tiene ciertas peculiaridades, aunque con frecuencia se percibe de forma característica cuando son más mayores. El lenguaje está afectado de forma constante y de forma aún más llamativa que el retraso motor, por lo que suelen acudir a consulta sobre los 15-18 meses y es ahí cuando suele sospecharse y consecuentemente diagnosticarse, aunque hay casos de diagnóstico más precoz, y desgraciadamente, mucho más tardío.
¿Existe algún procedimiento para detectarlo? ¿En qué consiste?
El retraso psicomotor y del lenguaje hacen saltar las alarmas. Junto al trastorno comunicativo de predominio expresivo, es frecuente la evitación del contacto ocular y ciertos rasgos que pueden observarse en el trastorno del espectro autista como las estereotipias o ciertas rigideces y obsesiones. Además, el fenotipo peculiar (cara alargada, frente amplia, mentón prominente y pabellones auriculares grandes y despegados) ayuda a intuir el diagnóstico. Ante dicha sospecha es conveniente realizar el estudio genético pertinente para diagnosticar el FXS.
¿Afecta más a los hombres o a las mujeres?
Al tener un cromosoma X extra para compensar, las mujeres padecen manifestaciones clínicas más leves, con un aspecto físico mucho menos evidente, salvo excepciones. Del mismo modo, los síntomas comentados previamente son casi siempre más leves, destacando cierta timidez, ansiedad, trastorno de atención y/o problemas de aprendizaje. Más de la mitad de las mujeres afectadas tienen un cociente intelectual normal.
¿Es una enfermedad hereditaria?
Efectivamente. El FXS es un trastorno ligado al cromosoma X. Es causado por una variante de pérdida de función en el gen de la ribonucleoproteína mensajera frágil X 1 (FMR1) ubicado en Xq27.3, lo que resulta en niveles disminuidos o ausentes de la fragile X mental retardation protein (FMRP). En más del 99% de los casos, la pérdida de función es causada por una expansión inestable de una repetición de trinucleótidos (esto es, de bases nitrogenadas que están repetidas más veces de lo que deberían). Por este motivo, existen dos niveles clínicamente significativos de la expansión. La mutación completa, con una expansión de más de 200 repeticiones, y la premutación, con una expansión más pequeña de entre aproximadamente 50 a 55 y 200 repeticiones, donde no se produce el fenotipo clásico del FXS, debido a que los niveles de FMRP son casi normales, pero que con el tiempo puede conducir a éste en una o más generaciones. Además, puede ocasionar en algunos individuos temblor y ataxia en la edad madura y en las mujeres un fallo ovárico prematuro.
¿Cuáles son los síntomas más frecuentes?
Las características clínicas del FXS varían según el estado de la mutación del gen FMR1 (mutación completa versus premutación), obviamente el sexo, el grado de metilación del gen y la magnitud del déficit de la proteína FMRP. Los varones con la mutación completa, además del retraso psicomotor (hipotonía junto a hiperlaxitud articular, particularmente de los pulgares, dedos y muñecas), del lenguaje y los rasgos "autistoides" (que la mayor parte de las veces no llegan a configurar un autismo completo), ya comentados, hay otros datos interesantes, como el agrandamiento testicular (volumen >25 mL después de la pubertad) después, al menos, de los 8 años de edad.
Casi la totalidad de los varones afectados presentan discapacidad intelectual con afectación moderada, aunque se ha identificado a un pequeño grupo con un funcionamiento intelectual límite. Más de la mitad presentan trastorno por déficit de atención / hiperactividad.
En ocasiones pueden existir crisis epilépticas, estrabismo y prolapso de la válvula mitral, entre otros síntomas.
¿Qué es el gen FMR1? ¿En qué se diferencia de otros?
El gen FMR1 se diferencia de otros en que se encarga de codificar la proteína FMRP. La elongación de las repeticiones CGG (las bases nitrogenadas que comentábamos) permite la hipermetilación de FMR1, lo que resulta en una transcripción deteriorada y una producción reducida de FMRP, dado que la metilación del ADN "apaga" la actividad del gen, impidiendo así la transcripción génica y por lo tanto de la proteína FMRP. Los niveles más bajos de ésta se relacionan con síntomas más graves en el SXF, dado que FMRP, muy presente en el sistema nervioso, se ensambla con otras proteínas formando complejos cruciales para el desarrollo adecuado de las neuronas y sus prolongaciones (sobre todo de las dendritas), procura una plasticidad correcta de las sinapsis (los contactos y conexiones entre neuronas) y también de sus canales iónicos (esto es, de la neurotransmisión).
¿Tiene cura?
Desgraciadamente no tiene cura. Sin embargo, su diagnóstico es importante para realizar precozmente una evaluación multidisciplinar que contemple las evaluaciones del desarrollo adecuadas (incluyendo el habla y el lenguaje, atención, habilidades académicas, resolución de problemas usando información visual, motricidad fina y gruesa) para la planificación de la estimulación del desarrollo temprano/intervención temprana del niño y los programas educativos especiales.
Es importante una evaluación conductual y psicológica para evaluar habilidades cognitivas, comportamiento adaptativo, problemas de concentración o atención, ansiedad, síntomas emocionales como la depresión, síntomas obsesivo-compulsivos y agresión o irritabilidad. Las intervenciones conductuales pueden ser efectivas para promover habilidades de afrontamiento y reducir comportamientos problemáticos (por ejemplo, agresión, comportamientos estereotipados, autolesiones). La intervención psicofarmacológica se individualiza según los síntomas (por ejemplo, falta de atención, hiperactividad, ansiedad, inestabilidad del estado de ánimo) y debe ser monitoreada de cerca.
Por supuesto, la evaluación médica es de sumo interés para detectar la presencia de problemas médicos asociados, incluidos problemas de alimentación, reflujo gastroesofágico, hipotonía, laxitud articular (incluyendo pies planos, hernia inguinal, escoliosis, etcétera), prolapso de la válvula mitral, crisis epilépticas, estrabismo y otitis media recurrente.
¿Por qué es importante detectarlo lo antes posible?
Por un lado, porque las terapias aumentan su efectividad cuando se implantan precozmente en el desarrollo del niño (de ahí el término "atención temprana"). Además, las familias de personas con FXS deben ser referidas a un genetista para una discusión detallada sobre la herencia de la mutación de FMR1 y la prueba de otros miembros de la familia, lo que debe realizarse pronto, por motivos obvios.
- 202227abr
El síndrome de kabuki (KS)
El síndrome de Kabuki (KS), es una patología con múltiples anomalías. Las más frecuentes: características faciales peculiares que remedan el maquillaje Kabuki -fisuras palpebrales largas y eversión de los párpados, que consiste en que éstos se ven como si se les hubiera dado la vuelta y se observa la parte interior, es decir, la parte que está en contacto con el ojo-, anomalías esqueléticas, engrosamiento de las yemas de los dedos y talla baja, asociadas a discapacidad intelectual. Pese a que se pensaba que era un síndrome muy raro, actualmente sabemos que afecta a cerca de una cada 30.000 personas aproximadamente, por lo que es una causa relativamente común de discapacidad intelectual, a tener siempre en cuenta, de ahí el interés del presente post.
El gen KMT2D, también conocido como MLL2, proporciona instrucciones para producir una enzima llamada metiltransferasa 2D específica de la lisina, que se encuentra en muchos órganos y tejidos del cuerpo. Esta enzima funciona modificando las histonas, agregándolas un grupo metilo (metilación), de este modo, las histonas metiltransferasas controlan la actividad de ciertos genes a nivel de empaquetamiento de la cromatina (la forma en la que se presenta el ADN en el núcleo celular). Así, la enzima codificada por KMT2D parece activar ciertos genes que son importantes para el desarrollo. Se han identificado diversas y múltiples mutaciones en el gen KMT2D, en personas con KS. El KDM6A, ligado al cromosoma X, puede ser responsable con menor frecuencia del KS, siendo más grave en varones. Existe la posibilidad de realizar el diagnóstico genético de esté síndrome en la práctica clínica, a través de una técnica denominada exoma.
Las anomalías estructurales en el KS pueden incluir lo siguiente:
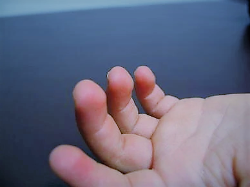
- Yemas de los dedos fetales persistentes (engrosamiento de las yemas de los dedos de las manos); se consideran una de las cinco manifestaciones cardinales del KS y, por lo tanto, se encuentran en una gran proporción de las personas afectadas.
- Oftalmológicas, incluyendo ptosis y estrabismo. Curiosamente, como resultado de la eversión del párpado inferior, los niños con KS pueden mostrar un lagrimeo excesivo, que generalmente no es un problema importante.
- Otológicas (una pista de diagnóstico potencialmente útil es que la mayoría de las personas con KS tienen orejas prominentes y en forma de copa. Los hoyos en el trago y en la región posterior de los pabellones auriculares también son relativamente comunes). La sordera neurosensorial es rara, aunque las otitis medias son comunes y a veces pueden producir pérdida de audición.
- Bucodentales: Labio y/o paladar hendido (un tercio de los niños). Anomalías dentales que incluyen dientes muy separados e hipodoncia (lo que significa que puede haber a veces incisivos superiores laterales ausentes, o incisivos inferiores ausentes, o molares superiores ectópicos y/o segundos premolares faltantes).
- Defectos cardíacos congénitos (cerca de la mitad de los casos, el más frecuente la coartación de aorta).
- Gastrointestinales, incluida la atresia anal. Las más frecuentes están relacionadas con hipotonía, mala coordinación oromotora y dificultades para tragar.
- Genitourinarias, incluyendo criptorquidia (testículo no descendido) en varones.
- Endocrinológicas, incluyendo telarquia prematura (aparición del botón mamario por primera vez en la mujer). En la adolescencia y la edad adulta, más de la mitad de las personas con KS desarrollan obesidad. La deficiencia del crecimiento posnatal es evidente a los 12 meses de edad. La falta de un crecimiento acelerado típico durante la pubertad exacerba la baja estatura. Pueden responder a tratamiento con hormona de crecimiento (GH). Otros hallazgos: hiperinsulinismo, insuficiencia suprarrenal, deficiencia combinada de hormona pituitaria, diabetes insípida, deficiencia franca de hormona de crecimiento, hipotiroidismo, disfunción ovárica primaria, verdadera pubertad precoz.
- Aumento susceptibilidad a enfermedades autoinmunes. No hay evidencia de aumento de cáncer.
- Neurológicas:
- Hipotonía, aunque la hiperlaxitud articular es la regla, lo que afecta al tono pasivo.
- Luxaciones o subluxaciones articulares, que afectan especialmente a las caderas, las rótulas y los hombros. No son infrecuentes, pero como en la mayoría de las condiciones con laxitud articular, este hallazgo mejora con la edad.
- Epilepsia o crisis epilépticas. No suelen ser de difícil control.
- Discapacidad intelectual, generalmente en el rango leve a moderado, en la mayoría de las personas; sin embargo, se han publicado informes de individuos con variantes patogénicas en KMT2D o KDM6A que tienen niveles de coeficiente intelectual superiores a 70. La mayoría de las personas con KS pueden hablar y caminar adecuadamente. No se ha identificado ningún perfil lingüístico específico. Sin embargo, todos los subdominios del lenguaje, incluidos la sintaxis, la morfología, la pragmática y la semántica, pueden verse afectados. En las pruebas neuropsiquiátricas formales, las personas con KS tienden a obtener mejores puntuaciones en las áreas de comprensión de vocabulario y memoria de trabajo y más bajas en las áreas de razonamiento no verbal y velocidad de procesamiento. Las personas con KS tienden a ser descritas como agradables y extrovertidas. En un subconjunto de individuos afectados existe TDAH. Rara vez se han informado otros trastornos: ansiedad, trastornos del espectro autista, trastornos de conducta, trastornos del sueño, etcétera.
El médico especialista, la mayor parte de las veces el Neuropediatra, debe decidir las pruebas a realizar en cada caso (ecografía renal/abdominal, EEG, interconsulta a cardiología, etc.) según los síntomas y signos detectados y/o esperados en cada paciente, de forma individualizada.
BIBLIOGRAFÍA:
Adam MP et al. Kabuki Syndrome. 2011 Sep 1 [Updated 2021 Jul 15]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022.
Banka S et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. European Journal of Human Genetics (2012) 20, 381–388.
Lepri FR. Clinical and Neurobehavioral Features of Three Novel Kabuki Syndrome Patients with Mosaic KMT2D Mutations and a Review of Literature. Int. J. Mol. Sci. 2018, 19, 82; doi:10.3390/ijms19010082.
- 202017nov
Retraso psicomotor en la infancia (III Parte)

Evaluación del desarrollo psicomotor (DPM)
"El Pediatra juega un papel trascendental en el diagnóstico precoz del retraso psicomotor".
Los controles periódicos de salud en los primeros años de vida, van a proporcionar al Pediatra un momento extraordinario para valorar el DPM del niño en cada momento, así como la evolución cognitiva, social, motora, entre otras esferas, que presentará en los primeros años de vida.
Los programas de seguimiento del niño sano permiten la evaluación transversal y evolutiva del niño. Para facilitar este seguimiento, el Pediatra puede hacer uso de diferentes escalas de desarrollo. Ninguna de las escalas de desarrollo tiene un correlato fiable con el cociente intelectual del niño mayor. Algunas de las que se usan son la Escalas de Desarrollo Infantil de Bayley –BSID-, que evalúa el desarrollo infantil desde el nacimiento hasta los 2,5 años o el Test de Screening de Desarrollo de Denver –DDST-. Posiblemente la escala más empleada. Se trata más de un registro o cuestionario que una escala de desarrollo. Valora cuatro áreas: motor-gruesa, motor-fina, personal-social y lenguaje. En sus diferentes versiones, registra el desarrollo en estas áreas hasta los 4 años de edad y el Test de Haizea-Llevant. Similar al DDST en su sistema de evaluación y estimación de áreas comprometidas. Elaborada de forma específica en niños españoles hasta los 4 años. La TABLA I muestra hallazgos que, típicamente suelen ser normales, durante la evolución de los niños.
Diagnóstico etiológico
"La historia clínica y la exploración física son los apartados más importantes en la evaluación etiológica del retraso psicomotor (RPM)".
Anamnesis
La historia clínica debe ser completa. Se debe recoger de forma detallada el desarrollo psicomotor del paciente, no sólo el desarrollo motor. En el caso de un estancamiento o involución, deben anotarse, entre otras, la edad de comienzo, las áreas afectadas, los síntomas acompañantes si existieron y las causas atribuidas por los padres u otros profesionales.
Dentro de este apartado se reflejarán igualmente los antecedentes personales, control del embarazo, infecciones, características del parto, edad gestacional, instrumentación, etcétera.
La recogida de datos relacionados con el periodo neonatal aporta de nuevo una información trascendental (Apgar, peso al nacimiento, cuidados neonatales…), el resultado de screening metabólico, la presencia de hipotonía o crisis en los primeros días de vida, los problemas respiratorios, y otros problemas.
En relación a los antecedentes personales posteriores no se obviarán aquellos trastornos o enfermedades que puedan tener relación con la situación a estudio: crisis epilépticas (con/sin fiebre), meningoencefalitis, traumatismos craneoencefálicos graves, cardiopatías, entre muchas otras. Finalmente se añadirán los antecedentes familiares. Debemos intentar obtener un árbol genealógico amplio, pero más centrado en padres, abuelos y hermanos, en el que se haga constar los posibles antecedentes llamativos.
Exploración física
Debe iniciarse por un examen general que incluya entre otros la exploración de rasgos dismórficos (TABLA II), aunque sean menores, el perímetro craneal (fundamental), el desarrollo ponderoestatural, las características cutáneas, el desarrollo óseo, la presencia de visceromegalias, y cualquier otro dato que nos llame la atención.
El Pediatra, y especialmente el neurólogo infantil, no deben temer la descripción de rasgos que le resultan inicialmente anormales. Igualmente, no debe obviarse la obtención de imágenes-fotografías del niño o familia, ante la presencia de rasgos pecuilares, o por otros motivos. En ocasiones una descripción fenotípica detallada es la que orienta el diagnóstico. En otras ocasiones el desarrollo ponderoestatural apoya un diagnóstico de sospecha; la anotación de la talla-peso-perímetro craneal desde edades precoces puede orientar al diagnóstico. La identificación de anomalías menores y mayores resulta trascendental en estos casos. Dentro del examen por sistemas, algunas alteraciones podrán sugerir la etiología de base. La presencia de trastornos pigmentarios cutáneos puede apuntar hacia trastornos neurocutáneos frecuentes como la neurofibromatosis (fig. 1), la hipomelanosis de Ito o la esclerosis tuberosa, u otros menos frecuentes como la enfermedad de von Hippel-Lindau o la incontinentia pigmenti (fig. 2); la fotosensibilidad podrá orientar hacia la enfermedad de Hartnup, el exantema malar hacia la homocistinuria...entre otras muchas posibilidades. Las alteraciones del cabello pueden ser relevantes (por ejemplo, en la enfermedad de Menkes, en el hipotiroidismo, etcétera). La presencia de hepatoesplenomegalia apuntará hacia mucopolisacaridosis, esfingolipidosis, glucogenosis, entre otras (TABLA III).
Tras abordar un examen físico completo, se debe proceder a la exploración neurológica igualmente completa, valorando cualquier signo focal presente, asimetrías en el examen (Fig. 3), no obviando el examen craneal, la impresión subjetiva del nivel cognitivo, entre otras variables. De igual modo, Se debe hacer un fondo de ojo, un examen auditivo y visual si se precisara. La audiometría convencional, la discriminación visual o auditiva, la campimetría por confrontación son medidas realizables en cualquier consulta pediátrica, si bien complejas en el niño de corta edad.
La historia clínica y la exploración física completa y minuciosa, serán las que deberán orientar al diagnóstico, y a la consecuente realización de las exploraciones complementarias oportunas. En éstas profundizaremos en la próxima revisión del blog.
Figura 1. Manchas color "café con leche" características de la neurofibromatosis tipo 1.
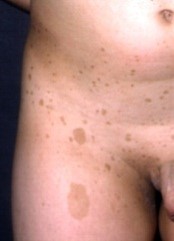
Figura 2. Lesiones características de la incontinentia pigmenti.
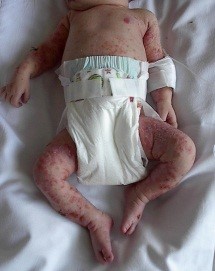
Figura 3. Marcha de inicio asimétrica de niño con hemiparesia izquierda.

TABLA I: VARIACIONES DE LA NORMALIDAD SIN CARÁCTER PATOLÓGICO
Pinza entre dedo pulgar y medio
Marcha de pie sin pasar por la fase de gateo
Desplazamiento sentado sobre nalgas o apoyando una rodilla y el pie de la otra extremidad o rodar sobre sí mismo
Marcha de puntillas primeras semanas o meses tras el inicio de la deambulación
Rotación persistente de la cabeza.
Retraso simple de la marcha con signo de ‘sentarse en el aire’
Tartamudeo fisiológico: entre los 2-4 años
Dislalias fisiológicas: hasta los 4-5 años
Otras: para neuropediatría
TABLA II. Malformaciones menores y mayores según localización (ejemplos)
Menores
Mayores
Cutáneas
Nevus
Hemangiomas
Manchas café con leche
Alopecia congénita
Hipertricosis
Craneales
Occipucio plano
Frente promiente
Craneosinostosis
Fístulas branquiales
Faciales
Hipertelorismo
Orificios nasales antevertidos
Boca en carpa
Orejas de implantación baja
Anoftalmia
Labio leporino
Atresia meato auditivo
Torácica
Tórax en tonel
Mamilas separadas
Malformaciones cardiovasculares
Abdominales
Hernia umbilical
Diastasis de rectos
Distensión abdominal
Malrotación o atresia intestinal
Onfalocele
Urogenital
Mínimo hipospadias
Teste en ascensor
Genitales ambigüos
Criptorquidia
Epispadias
Esqueléticas
Cubitus valgo
Genu recurvatum
Fosita sacra
Pie equinovaro
Hemivértebras
Polidactilia
SNC
Displasias corticales
Meningocele
TABLA III. Afectación de diferentes órganos o sistemas según trastorno metabólico
Afectación hepática y/o esplénica
Con hepatoesplenomegalia:
Esfingolipidosis, mucolipidosis, mucopolisacaridosis, trastornos peroxisomales, galactosemia…
Con ictericia y fallo hepático:
Enfermedad de Wilson, enfermedad de Niemann-Pick C, síndrome de Alpers, galactosemia…
Cardiopatía
Glucogenosis tipo 2, enfermedad de Friedreich, enfermedad de Refsum, enfermedad de Fabry, mucopolisacaridosis, homocistinuria…
Nefropatía
Síndrome de Lowe, enfermedad de Zellweger, enfermedad de Fabry, enfermedad de Lesch-Nyhan, galactosemia, acidemia isovalérica…
Afectación esquelética
Mucolipidosis, mucopolisacaridosis, sialidosis, enfermedad de Zellweger, enfermedad de Lowe, enfermedad de Refsum…
Afectación cutánea
Enfermedad de Hartnup, síndrome de Cockayne, xeroderma pigmentoso, fucosidosis, déficit de biotinidasa…
Anomalías hematológicas
Anemia:
Enfermedad de Gaucher, enfermedad de Fabry…
Trombocitopenia:
Acidemia isovalerica, acidemia propiónica, enfermedad de Wilson…
Afectación respiratoria
Enfermedad de Menkes, glucogenosis tipo II, enfermedad de Farber…
Afectación digestiva
Abetalipoproteinemia, MELAS, porfiria aguda intermitente…
- 20203nov
Retraso psicomotor en la infancia (II Parte)

Clasificación etiológica
"Un retraso psicomotor no siempre es patológico o anormal, pero puede ser también la antesala de graves problemas del desarrollo físico y cognitivo del niño"
1. Variante de la normalidad.
Los márgenes de la normalidad para numerosos hitos son amplios. En ocasiones, especialmente en RPM parciales, encontramos pacientes completamente sanos, que se "escapan" de los márgenes señalados como "normales" para la población a estudio, los signos más frecuentes aparecen en la TABLA III. Por ejemplo, un tercio de los niños no gatea nunca, por lo que es un signo más "tranquilizador" en su presencia que de "alarma" en su ausencia.
Dos circunstancias especiales en este sentido son el recién nacido prematuro (RNPT) y el niño ingresado-encamado. El RNPT alcanzará los hitos lógicos del DPM más tarde que los demás; para valorar la normalidad del desarrollo en estos niños, deberá emplearse la edad corregida, es decir la edad que el niño tendría si hubiera nacido en la fecha prevista del parto (edad corregida= "edad cronológica medida en semanas o meses"-"número de semanas o meses de prematuridad"). Esta corrección es especialmente necesaria en los primeros 24 meses. Por otro lado, la prematuridad es un factor de riesgo para los problemas del desarrollo y el aprendizaje, por lo que el DPM deberá ser vigilado estrechamente. El niño ingresado o encamado durante tiempos largos durante el 1º-2º año de vida, puede igualmente mostrar un leve retraso o estancamiento del desarrollo motor. En estos niños se puede sumar el RPM a déficit asociados en el desarrollo por la patología que justificó el ingreso hospitalario.
2. Hipoestimulación.
Los niños pobremente estimulados o institucionalizados pueden mostrar un claro RPM en los primeros meses de la vida. Esta circunstancia es generalmente normalizable. Sin embargo, cuando la hipoestimulación es severa y mantenida, como sucede en niños adoptados del este de Europa, puede justificar, junto a otros factores de riesgo, futuros problemas del neurodesarrollo.
3. Déficit neurosensorial
Los problemas sensoriales, especialmente auditivos o visuales, pueden ser causa de un RPM. Es habitual que la patología auditiva severa se asocie con retrasos del lenguaje, la comunicación, e incluso con conductas de aislamiento que pueden recordar trastornos generalizados del desarrollo. Por ello deben descartarse en todos los casos, bien por audiometría, bien por potenciales evocados auditivos del tronco encefálico (PEATE) si el niño no colabora o es muy pequeño. Los niños con trastornos auditivos pueden mostrar un desarrollo del lenguaje normal los primeros 6 meses de vida (ruidos, risas, balbuceos…) con interrupción del mismo por ausencia de feedback ambiental. Es excepcional que la hipoacusia leve uni o bilateral justifique un verdadero retraso del lenguaje; del mismo modo, no debe justificarse el retraso del lenguaje a otitis recurrentes.
Los problemas visuales pueden igualmente asociarse a problemas de la coordinación, manipulación… Estos generalmente están relacionados con el componente visual-sensorial, no con el motor; un estrabismo o nistagmus puede ser un signo de un trastorno neurológico de base, pero no la causa de un RPM
4. Anticipación de un trastorno específico del desarrollo
Los trastornos del desarrollo de la coordinación y los trastornos de la comunicación, tienden a anticiparse por RPM con afectación predominantemente motora y del lenguaje respectivamente.
5. Anticipación de un trastorno motor
La parálisis cerebral infantil (PCI) tiende a manifestarse en los primeros 18-24 meses de vida por un RPM global o predominantemente motor. Aunque PCI se define como un trastorno motor, crónico, de comienzo precoz y no progresivo, las manifestaciones clínicas pueden ser cambiantes y más invalidantes durante el desarrollo del niño. Puede además acompañarse de problemas sensoriales (visuales hasta en el 50% y auditivos hasta en el 15% de los casos), epilepsia (25-35%) que puede condicionar el propio desarrollo global o discapacidad intelectual (DI) -retraso mental- (hasta en el 50% de los niños).
En este apartado debemos incluir las miopatías, tanto las congénitas como las distrofias musculares, que pueden manifestarse con carácter estático o progresivo respectivamente, y a veces acompañadas de retraso cognitivo. Igualmente, no deben obviarse otras enfermedades como la atrofia muscular espinal que se manifestará en los primeros meses de vida, o algunas neuropatías genéticas, que podrán hacerlo en los 3-4 primeros años, en forma de retraso motor, hipotonía o torpeza.
6. Anticipación de una discapacidad intelectual –DI- (retraso mental)
Generalmente la mayoría de los pacientes con DI (este término ha ido sustituyendo al de retraso mental), han tenido al menos cierto RPM. En ocasiones, las DI leves se anticipan por leves RPM o RPM parciales, que pasan desapercibidos para la familia o el médico.
Es un trastorno plurietiológico; habitualmente de causa genética. Estos últimos años el diagnóstico etiológico ronda el 50% de los casos leves y el 80% de los graves gracias a los espectaculares avances en el diagnóstico genético estos últimos 3-4 años.
Es frecuente que los pacientes con DI asocien otros problemas neurológicos que contribuyen de forma desfavorable en el DPM. Algunos estudios refieren la asociación a encefalopatías motoras en el 7% de los pacientes, epilepsia en el 10%, alteraciones neurosensoriales en el 7% o autismo en el 2-3%. Estas asociaciones se muestran más intensas cuanto menor es el CI.
7. Anticipación de un trastorno del espectro autista (TEA)
Caracterizado eminentemente por una alteración de la socialización, la comunicación y un patrón de intereses restringidos y comportamientos estereotipados, se puede manifestar con un desarrollo lento o atípico. Estos problemas pueden acompañarse de cierta torpeza o hipotonía en los primeros meses de vida (a menudo debido a la causa genética subyacente del trastorno), pero suele expresarse en los primeros meses/años de vida con una alteración cualitativa y/o cuantitativa del lenguaje y ser indiferenciable inicialmente de un trastorno específico del lenguaje (TEL) con afectación de la comprensión del mismo. En estos casos lo más importante es derivar al neuropediatra e instaurar una intervención precoz adecuada mientras se realizan las pruebas complementarias oportunas. La ausencia de un diagnóstico específico no puede demorar la derivación de un niño con sospecha de TEA/TEL a un centro de atención temprana especialista en trastornos de la comunicación. Separar este grupo de los anteriores se muestra complejo, dada de nuevo su comorbilidad con otros trastornos o enfermedades. Hasta el 90% de los casos de TEA grave pueden tener DI, y epilepsia hasta en la mitad de los casos (especialmente si tienen DI), alteraciones visuales o auditivas leves que pueden condicionar el DPM, etcétera.
Tabla. Prevalencia de las principales causas del RPM global o parcial.
Sordera
0,1%
Ceguera
1,5-6/10000
Trastorno del desarrollo de la coordinación
6%
Trastorno de la comunicación
4-6%
Parálisis cerebral infantil
0,2%
Retraso mental (discapacidad intelectual)
1%
Autismo
1%
Sobre este blog
Blog sobre los temas relacionados con la neuropedciatría: déficit de atención, hiperactividad, epilepsia, cefaleas, tics, encefalitis, problemas escolares, etc.

Archivo del blog
 2.026
2.026
- 2.025
- Diciembre
- Noviembre
- Octubre
- La niebla del diagnóstico: por qué el cannabis complica el TDAH y la psicosis juvenil de formas inesperadas
- Cómo influye el uso de cannabis en la adolescencia sobre la maduración del cerebro y las capacidades cognitivas
- Efectos del cannabis en la salud física del adolescente
- TDAH y emociones fuera de control: entender la desregulación emocional como un síntoma central del trastorno
- Vértigo en niños: ¡calma, padres! Entendiendo los mareos de nuestros pequeños
- Septiembre
- Agosto
- Julio
- Junio
- Mayo
- Abril
- Marzo
- Enero
- 2.024
- 2.023
- 2.022
- 2.021
- 2.020
- 2.019
- 2.018
- Diciembre
- Noviembre
- Octubre
- Julio
- Mayo
- Abril
- Febrero
- Recomendaciones generales y específicas para el trastorno por déficit de atención/hiperactividad -TDAH- (III). Recomendaciones específicas.
- El Dr. Daniel Martín Fernández-Mayoralas ganador del “Concurso de Casos Clínicos sobre el abordaje farmacológico de pacientes con TDAH" organizado por el Grupo Saned
- Enero
- 2.017
- 2.016
Últimas entradas
- La psicología de prescribir psicofármacos en niños y adolescentes: más allá de la receta
- El neuropediatra y los trastornos del sueño: ¿cuándo y por qué consultarlo?
- Cuando el mundo es demasiado intenso: sensibilidad sensorial y ansiedad en el autismo
- Terapias psicológicas para el TEA de alto rendimiento (Grado 1): guía práctica para padres
- Esofagitis eosinofílica en neurología y psiquiatría infantil: un diagnóstico olvidado



La finalidad de este blog es proporcionar información de salud que, en ningún caso sustituye la consulta con su médico. Este blog está sujeto a moderación, de manera que se excluyen de él los comentarios ofensivos, publicitarios, o que no se consideren oportunos en relación con el tema que trata cada uno de los artículos.
Quirónsalud no se hace responsable de los contenidos, opiniones e imágenes que aparezcan en los "blogs". En cualquier caso, si Quirónsalud es informado de que existe cualquier contenido inapropiado o ilícito, procederá a su eliminación de forma inmediata.
Los textos, artículos y contenidos de este BLOG están sujetos y protegidos por derechos de propiedad intelectual e industrial, disponiendo Quirónsalud de los permisos necesarios para la utilización de las imágenes, fotografías, textos, diseños, animaciones y demás contenido o elementos del blog. El acceso y utilización de este Blog no confiere al Visitante ningún tipo de licencia o derecho de uso o explotación alguno, por lo que el uso, reproducción, distribución, comunicación pública, transformación o cualquier otra actividad similar o análoga, queda totalmente prohibida salvo que medie expresa autorización por escrito de Quirónsalud.
Quirónsalud se reserva la facultad de retirar o suspender temporal o definitivamente, en cualquier momento y sin necesidad de aviso previo, el acceso al Blog y/o a los contenidos del mismo a aquellos Visitantes, internautas o usuarios de internet que incumplan lo establecido en el presente Aviso, todo ello sin perjuicio del ejercicio de las acciones contra los mismos que procedan conforme a la Ley y al Derecho.



© 2026 Quirónsalud - Todos los derechos reservados
